Miopatías Inflamatorias Idiopáticas
INTRODUCCIÓN
Las miopatías inflamatorias idiopáticas (MII) son un grupo heterogéneo de enfermedades autoinmunes musculares de causa desconocida. Su característica principal es la debilidad muscular proximal y simétrica progresiva, disminución de la resistencia muscular y fatiga.
Pueden presentarse de forma aislada, asociada a otras enfermedades autoinmunes, neoplasias y más raro a una infección.
Aunque clásicamente incluyen la dermatomiositis (DM) y la polimiositis (PM), otras enfermedades como la miositis con cuerpos de inclusión (MCI) o las miopatías necrosantes inmunomediadas se han incorporado en este grupo en los últimos años.
Las miopatías inflamatorias idiopáticas son enfermedades sistémicas y se acompañan de fenómenos de autoinmunidad, especialmente la presencia de autoanticuerpos.
EPIDEMIOLOGÍA
Las MII son enfermedades poco frecuentes. La incidencia anual media global oscila entre 2.2 y 7.7 casos por millón de habitantes.
Pueden aparecer a cualquier edad, aunque, predomina claramente por encima de los 50 años, especialmente entre los 55 a 65 años. También se puede presentar en la edad infantil.
En general la prevalencia de las MII es mayor en mujeres, aunque existe un claro predominio de MCI en hombres.
Se ha encontrado asociación de las miopatías con otras enfermedades autoinmunes especialmente en pacientes con enfermedad mixta el tejido conectivo (EMTC), esclerodermia y lupus.
En los casos de PM y DM, la frecuencia de neoplasia está aumentada, con mayor predominio en pacientes con DM.
FORMAS CLÍNICAS
DERMATOMIOSITIS
Su característica distintiva es la presencia de manifestaciones cutáneas típicas, que acompañan o, más frecuentemente, preceden a la aparición de la debilidad muscular.
Las lesiones principales se describen a continuación:
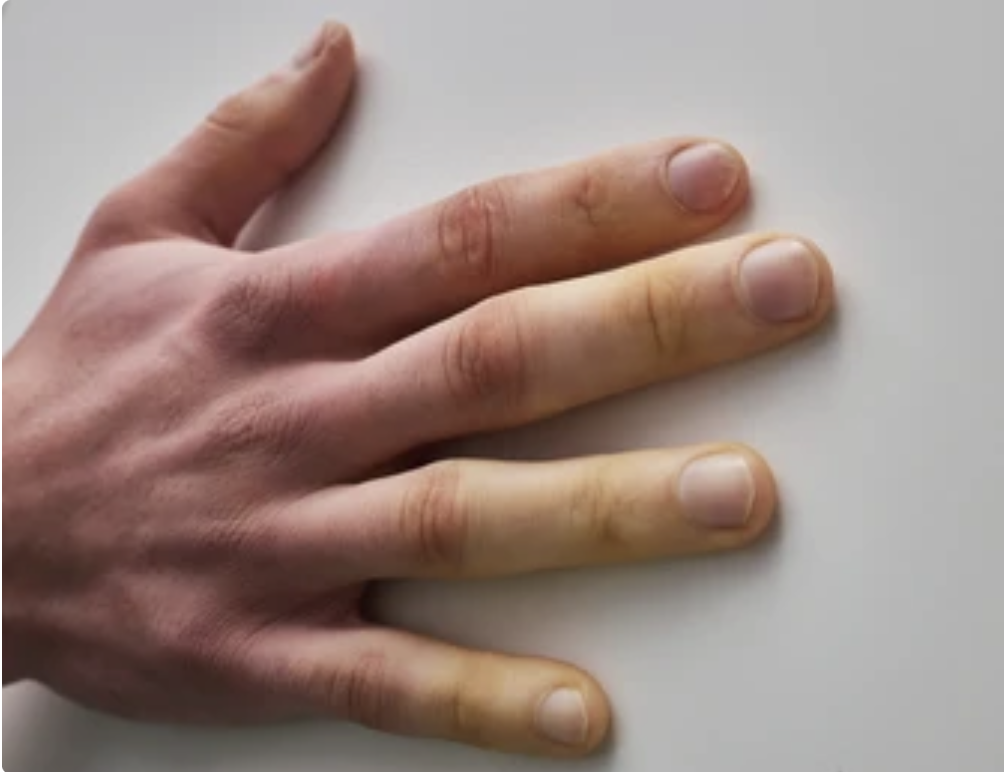
- Signo de Gottron: es una erupción eritematosa simétrica, que aparece en la zona de los nudillos de las manos. También puede observarse sobre las rodillas y codos, con un aspecto parecido a la psoriasis.
- Heliotropo: es una erupción violácea sobre los párpados.
- Lesiones en chal: lesiones eritematosas en zonas expuestas en “chal” o en “V” del escote, con componente de fotosensibilidad (empeora con el sol).
- Manos de mecánico: se presentan con hiperqueratosis en el área lateral de los dedos. Se asocian a la presencia de síndrome antisintetasa.
- Alteraciones periungueales: estas se presentan como eritema periungueal y cambios en la capilaroscopía similares a otras conectivopatías, con disminución de capilares y dilataciones.
- Calcinosis: se refiere al depósito de calcio en la piel que aparece especialmente en los niños con DM.
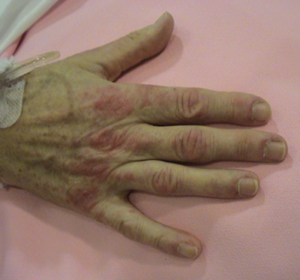
Eritema en nudillos : signo de gottron
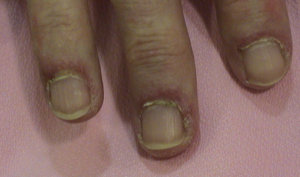
Eritema periungueal, el cual puede mostrar alteraciones vasculares en la capilaroscopía
Las manifestaciones cutáneas acostumbran a preceder a la debilidad muscular, que es, generalmente de presentación subaguda, afectando a la musculatura proximal de las extremidades, cintura pelviana y escapular, cervical, y, en ocasiones puede afectar los músculos de la deglución y respiratorios. El grado de debilidad muscular es variable.
Ocasionalmente, las manifestaciones cutáneas no se acompañan de debilidad muscular, esta es mínima, o se encuentra la presencia de enzimas musculares elevados ( Creatina kinasa : CK ) , alteraciones electrofisiológicas o de imagen. Para estos casos se ha acuñado el término dermatomiositis amiopática o dermatomiositis sin miositis. En estas ocasiones, si se realiza una biopsia muscular, se puede demostrar la presencia de enfermedad.
La frecuencia de asociación concomitante de neoplasia se encuentra aumentada llegando hasta el 25% de los casos durante los primeros 3-5 años. Puede asociarse también en los casos de dermatomiositis amiopática. El cáncer de mama y de ovario son los más frecuentes en las mujeres y los de pulmón y próstata en hombres.
POLIMIOSITIS
La presentación clínica característica es la aparición de debilidad muscular insidiosa, proximal, simétrica, con frecuencia afecta a musculatura cervical, con empeoramiento progresivo entre semanas a meses. Generalmente respeta la musculatura bulbar y facial, aunque en determinados casos puede acompañarse de disfagia e incluso insuficiencia respiratoria.
La PM debería considerarse un diagnóstico de exclusión ante la presencia de un cuadro de debilidad muscular de cinturas con presentación subaguda de inicio en la edad adulta.
Como entidad clínica aislada es poco común dado que, el cuadro clínico característico puede encontrarse en el contexto de enfermedades sistémicas autoinmunes o del tejido conectivo, como la esclerodermia o el lupus
Los síntomas sistémicos como fiebre, pérdida de peso, artralgias o fenómeno de Raynaud pueden estar presentes en los casos de asociación con enfermedades del tejido conectivo. Otras manifestaciones no musculares son la presencia de síntomas respiratorios que pueden ser consecuencia de la afectación de la musculatura respiratoria, pero, generalmente, derivan de la presencia enfermedad pulmonar intersticial asociada, que puede afectar hasta a la mitad de los pacientes.
MIOSITIS POR CUERPOS DE INCLUSIÓN
Clásicamente ha sido clasificada en el grupo de miopatías inflamatorias idiopáticas junto con la PM y la DM. Sin embargo, además de las características patológicas particulares, desde un punto de vista clínico, presenta más diferencias que similitudes con éstas.
La debilidad muscular puede ser asimétrica y distal, incluso puede afectar a músculos faciales, aunque no afecta la musculatura oculomotora. No es inusual la asociación de disfagia y que durante la evolución se vea afectada la musculatura distal de extremidades inferiores. En el 40% de los casos se acompaña de dolor muscular.
Afecta con mayor frecuencia a hombres y se presenta de manera insidiosa con un curso lentamente progresivo, de manera casi invariable en pacientes mayores de 40 años y se considera que es la miopatía más frecuente en pacientes mayores de 50 años.
MIOPATÍAS NECROTIZANTES INMUNOMEDIADAS
Se trata de un grupo heterogéneo de entidades que comparten una presentación clínica común en forma de debilidad muscular asociada a características específicas en el estudio del tejido muscular.
La debilidad se presenta a nivel de la musculatura proximal de extremidades, de manera simétrica, instaurándose, generalmente, de manera subaguda, aunque puede presentarse, también, de manera insidiosa y lentamente progresiva.
No es infrecuente la asociación de disfagia o disartria en casos de debilidad muy grave, sintomatología respiratoria, o manifestaciones cutáneas, articulares y fenómeno de Raynaud. Las mialgias son infrecuentes o leves, pero la debilidad puede acompañarse de pérdida de peso significativa. En casos en que ésta se acompañe de fiebre o sudoración nocturna debe buscarse una neoplasia subyacente.
Se han clasificado en cinco formas diferentes en función de determinadas características de los infiltrados inflamatorios, tinciones inmunohistoquímicas, características clínicas o presencia de auto-anticuerpos.
DIAGNÓSTICO
El diagnóstico de estas enfermedades se basa en los hallazgos de la historia clínica , la exploración física y los pruebas complementarias destacando:
LABORATORIO
- Enzimas musculares: Creatinkinasa (CK) La elevación de las enzimas musculares está en relación con la presencia de fibras musculares lesionadas.
- Otros : GOT, GPT y LDH se encuentran elevadas si existe lesión muscular, pero también en enfermedades hepáticas.
- Anticuerpos: La frecuencia de anticuerpos varían según la enfermedad, pero más del 50% de los pacientes con MII los presentan.
- Anticuerpos antinucleares (ANA) Con inmunofluorescencia indirecta, entre el 50%-80% de los pacientes con MII presentan ANA positivos “moteados”.
- Anticuerpos específicos de miositis (AEM) Se refieren a anticuerpos en los que la mayor parte de los pacientes tienen miositis, PM o DM. Estos son => Anti-Jo-1 o antisintetasa, Anti Mi-2, Anti-SRP.
- Anticuerpos asociados a miositis (AAM). Estos anticuerpos con frecuencia se encuentran asociados a cuadros de síndrome de solapamiento en los que la miositis no es el dato fundamental. Pueden ser de utilidad para clasificación diagnóstica de los pacientes. Los más importantes son: anti-PM-Scl, anti-U1RNP, anti-Ku, anti-Ro.
ELECTROFISIOLOGÍA
- El estudio electrofisiológico (electromiografía) es característico y sugestivo de MII en el 50% de los pacientes.
BIOPSIA MUSCULAR
- Puede ser necesaria para el diagnóstico definitivo de la enfermedad muscular.
RESONANCIA MAGNÉTICA (RM)
- La RM es útil para diferenciar la presencia de actividad de la miositis del daño residual.
CAPILAROSCOPÍA
- Es una prueba no invasiva que permite visualizar la afectación endotelial a través del lecho ungueal.
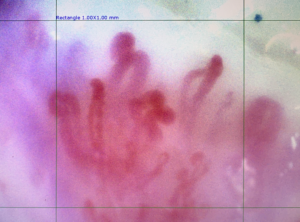
Imagen de capilaroscopía con patrón de neoangiogénesis (neoformación vascular) en paciente con dermatomiositis ( Imagen Dra Jeria)
TRATAMIENTO
El objetivo principal del tratamiento es el control de la inflamación muscular, mejorar la debilidad muscular, evitar la incapacidad y las complicaciones.
En general, el tratamiento se basa en el uso de corticoides asociado o no a inmunosupresores como metotrexato, azatioprina, micofenolato etc.
Otras medidas generales de importancia incluyen: iniciar un tratamiento de fisioterapia supervisado, protección solar en los pacientes con DM y medidas para prevenir la osteoporosis como el uso de bifosfonatos y suplementos de calcio y vitamina D.
Es necesario un control estrecho de estos pacientes para anticiparse al desarrollo de daño irreversible.
Gracias por leer hasta el final, te ha parecido interesante ?
Nos vemos en otra publicación de “Mi reumatóloga”
BIBLIOGRAFÍA
- Schmidt J. Current Classification and Management of Inflammatory Myopathies. J Neuromuscul Dis. 2018;5(2):109-129. doi: 10.3233/JND-180308.
- Lundberg IE, Miller FW, Tjärnlund A, Bottai M. Diagnosis and classification of idiopathic inflammatory myopathies. J Intern Med. 2016 Jul;280(1):39-51. doi: 10.1111/joim.12524.
- Stenzel W, Goebel HH, Aronica E. Review: immune-mediated necrotizing myopathies–a heterogeneous group of diseases with specific myopathological features. Neuropathol Appl Neurobiol 2012;38:632-646. doi:10.1111/j.1365-2990.2012.01302.x